Cosa sono le mucopolisaccaridosi
Le mucopolisaccaridosi (MPS) sono un gruppo eterogeneo di malattie ereditarie causate dalla mancanza degli enzimi lisosomiali necessari per la degradazione dei glicosaminoglicani (GAG o mucopolisaccaridi). I GAG sono molecole coinvolte in numerosi processi di regolazione e comunicazione cellulare che vengono periodicamente degradati all’interno di strutture cellulari, dette lisosomi. A causa della carenza di uno specifico enzima si può verificare un accumulo di un determinato tipo di GAG.
I glicosaminoglicani (chiamati anche GAG o mucopolisaccaridi) sono molecole coinvolte in numerosi processi di regolazione e comunicazione cellulare e vengono periodicamente degradati all’interno dei lisosomi. A causa della carenza di uno specifico enzima si può verificare un accumulo di un determinato GAG.
Le funzioni dei glicosaminoglicani sono determinate dalla loro struttura molecolare. In passato i GAG si ritenevano utili ai soli fini dell’idratazione della cellula e della sua struttura, mentre al giorno d’oggi si ha la consapevolezza che questi polisaccaridi interagiscono con un'ampia varietà di proteine e molecole di segnalazione nell'ambiente cellulare e ne modulano l'attività.
Nello specifico:
• Regolano la crescita e la proliferazione cellulare.
• Promuovono l’adesione cellulare.
• Partecipano all’anticoagulazione e alla riparazione delle ferite.
• Rappresentano funzioni di sostegno e protezione della maggior parte dei tessuti.
Le mucopolisaccaridosi sono un gruppo di malattie genetiche rare che si manifestano in età infantile e sono causate da un accumulo lisosomiale delle molecole appartenenti alla famiglia dei mucopolisaccaridi (GAG) in tutte le cellule e tessuti dei pazienti.
Le mucopolisaccaridosi si caratterizzano per la carenza di enzimi lisosomiali necessari per il metabolismo dei glicosaminoglicani; dal momento che quest’ultimi non si degradano, essi continuano ad accumularsi durante lo sviluppo, causando una forte disfunzione cellulare e, infine, il decesso del paziente.
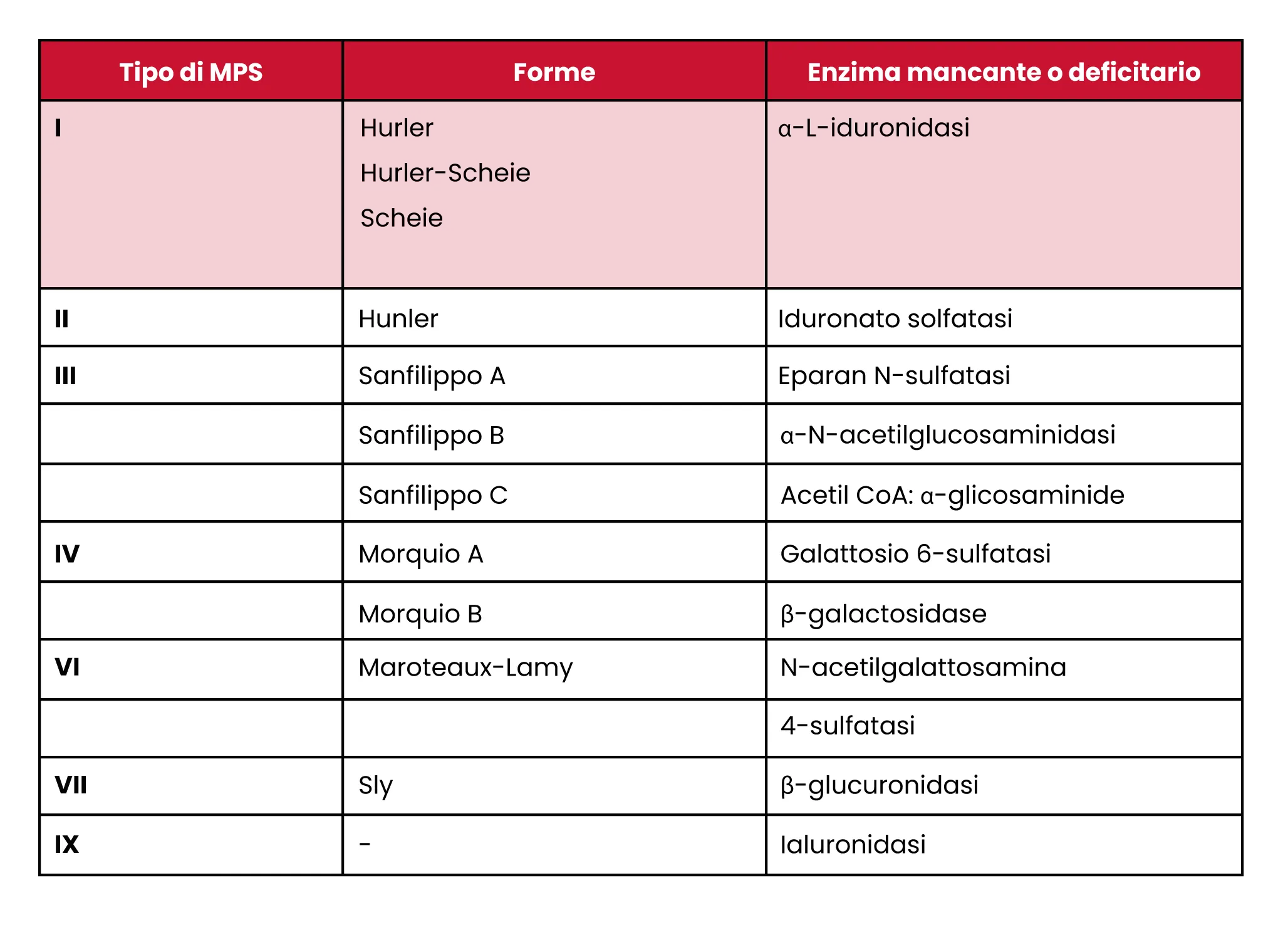
In base all’enzima disfunzionale e alla conseguente alterazione del metabolismo dei glicosaminoglicani interessati nei sistemi di organi, ad oggi le mucopolisaccaridosi vengono classificate in sette tipi:
I sintomi della mucopolisaccaridosi si sviluppano nel tempo in base alla gravità della malattia, mentre il danno causato agli apparati e agli organi coinvolti peggiora progressivamente.
MPS I
• Sindrome di Hurler: i sintomi si manifestano durante il primo anno di vita e tale malattia rappresenta la forma più grave di Mucopolisaccaridosi di tipo I. Il peggioramento è rapidamente riscontrabile a discapito del normale sviluppo fisico, emotivo e sociale, così come del linguaggio e dell’apprendimento. Il decesso è solitamente previsto entro la prima decade di vita.
• Sindrome di Scheie: si manifesta dopo i 5 anni d’età. Si tratta di una forma più lieve di questo tipo di malattia da accumulo lisosomiale; peggiora più lentamente ed è possibile una normale aspettativa di vita fino alla quinta o sesta decade, considerando che apprendimento e statura sono nei limiti di norma.
• Sindrome di Hurler-Scheie: si manifesta tra i 3 e gli 8 anni. Si posiziona come via di mezzo tra le prime due forme di MPS I, ma peggiora piuttosto rapidamente; non sempre presenta ritardo nello sviluppo (bassa statura) e l’aspettativa di vita arriva alla seconda o terza decade.
MPS II
• Sindrome di Hunter: i sintomi si manifestano tra i 2 e i 4 anni e sono simili a quelli della sindrome di Hurler, con l’eccezione che non si riscontrano problemi alla vista. La forma più grave presenta disfunzioni cognitive e un’aspettativa di vita intorno ai 15 anni.
MPS III
• Sindrome di Sanfilippo: si manifesta tra i 2 e i 4 anni, inizialmente con accentuati disturbi comportamentali e, a seguire, con problemi legati al linguaggio e all’apprendimento. In media l’aspettativa di vita non supera la seconda decade.
MPS IV
• Sindrome di Morquio: i sintomi si presentano tra il primo e il quarto anno di età e sono simili a quelli della sindrome di Hurler, ma con una più accentuata disostosi multipla.
MPS VI
• Sindrome di Maroteaux-Lamy: la progressione della malattia inizia lentamente per poi aggravarsi in modo più rapido a livello articolare e scheletrico, senza però sfociare in una disabilità intellettuale. La vita ha una durata media di due o tre decadi.
MPS VII
• Sindrome di Sly: ha diversi gradi di gravità, ma si manifesta attraverso sintomi molto simili a quelli della sindrome di Hurler.
MPS IX:
• Carenza di Ialuronidasi: si presenta sin dai primi 6 mesi di vita con lineamenti non nella norma e possibili tumori nella zona delle articolazioni; non manifesta però disabilità cognitiva.
La non specificità dei sintomi e la loro manifestazione progressiva rende la diagnosi complicata. Quando si riscontrano dei disturbi all’apparato muscolo-scheletrico, gastrointestinale, cardiaco, visivo o uditivo, è possibile che tali sintomi non vengano immediatamente connessi alla diagnosi di MPS, dal momento che si tratta di una patologia complessa che coinvolge più organi.
Per eliminare qualsiasi dubbio e diagnosticare definitivamente una tra le malattie da accumulo lisosomiale, è utile ricercare nelle urine il dosaggio di glicosaminoglicani (GAG). Successivamente, per confermare e identificare il tipo di MPS è importante eseguire analisi biochimiche più dettagliate.
Il trattamento delle mucopolisaccaridosi si basa su un alleviamento dei sintomi e una compensazione enzimatica in grado di agire sulla causa della malattia, rallentare la sua progressione e migliorare la qualità della vita. Alcune mucopolisaccaridosi possono essere trattate con la terapia enzimatica sostitutiva (ERT), tramite iniezione endovenosa, in modo tale da colmare la mancanza dell’enzima in questione, oppure tramite un trapianto di midollo osseo o di cellule staminali ematopoietiche. Quest’ultimo avviene grazie alla disponibilità di un donatore e attraverso un intervento chirurgico.
La non specificità dei sintomi e la loro manifestazione progressiva rende la diagnosi complicata. Quando si riscontrano dei disturbi all’apparato muscolo-scheletrico, gastrointestinale, cardiaco, visivo o uditivo, è possibile che tali sintomi non vengano immediatamente connessi alla diagnosi di MPS, dal momento che si tratta di una patologia complessa che coinvolge più organi.
Per eliminare qualsiasi dubbio e diagnosticare definitivamente una tra le malattie da accumulo lisosomiale, è utile ricercare nelle urine il dosaggio di glicosaminoglicani (GAG). Successivamente, per confermare e identificare il tipo di MPS è importante eseguire analisi biochimiche più dettagliate.
Il trattamento delle mucopolisaccaridosi si basa su un alleviamento dei sintomi e una compensazione enzimatica in grado di agire sulla causa della malattia, rallentare la sua progressione e migliorare la qualità della vita. Alcune mucopolisaccaridosi possono essere trattate con la terapia enzimatica sostitutiva (ERT), tramite iniezione endovenosa, in modo tale da colmare la mancanza dell’enzima in questione, oppure tramite un trapianto di midollo osseo o di cellule staminali ematopoietiche. Quest’ultimo avviene grazie alla disponibilità di un donatore e attraverso un intervento chirurgico.
Leggi anche:
- Conoscere la MPS I e le mucopolisaccaridosi
- Trasmissione ed ereditarietà della MPS I
Bibliografia
Fecarotta S et al. Int J Mol Sci. 2020 Apr 4;21(7):2515.
Khan SA et al. Mol Genet Metab. 2017 Jul;121(3):227-240.
Linhardt RJ, Toida T. Acc Chem Res. 2004 Jul;37(7):431-8.
Sun A. Ann Transl Med. 2018 Dec;6(24):476.
MAT-IT-2100680