Conoscere l’ASMD

La Malattia di Niemann-Pick (NPD) fa parte di un gruppo di malattie metaboliche ereditarie caratterizzate da un eccessivo e dannoso accumulo di lipidi (grassi) in diversi organi - in particolare di cervello, milza, fegato, polmoni e midollo osseo.
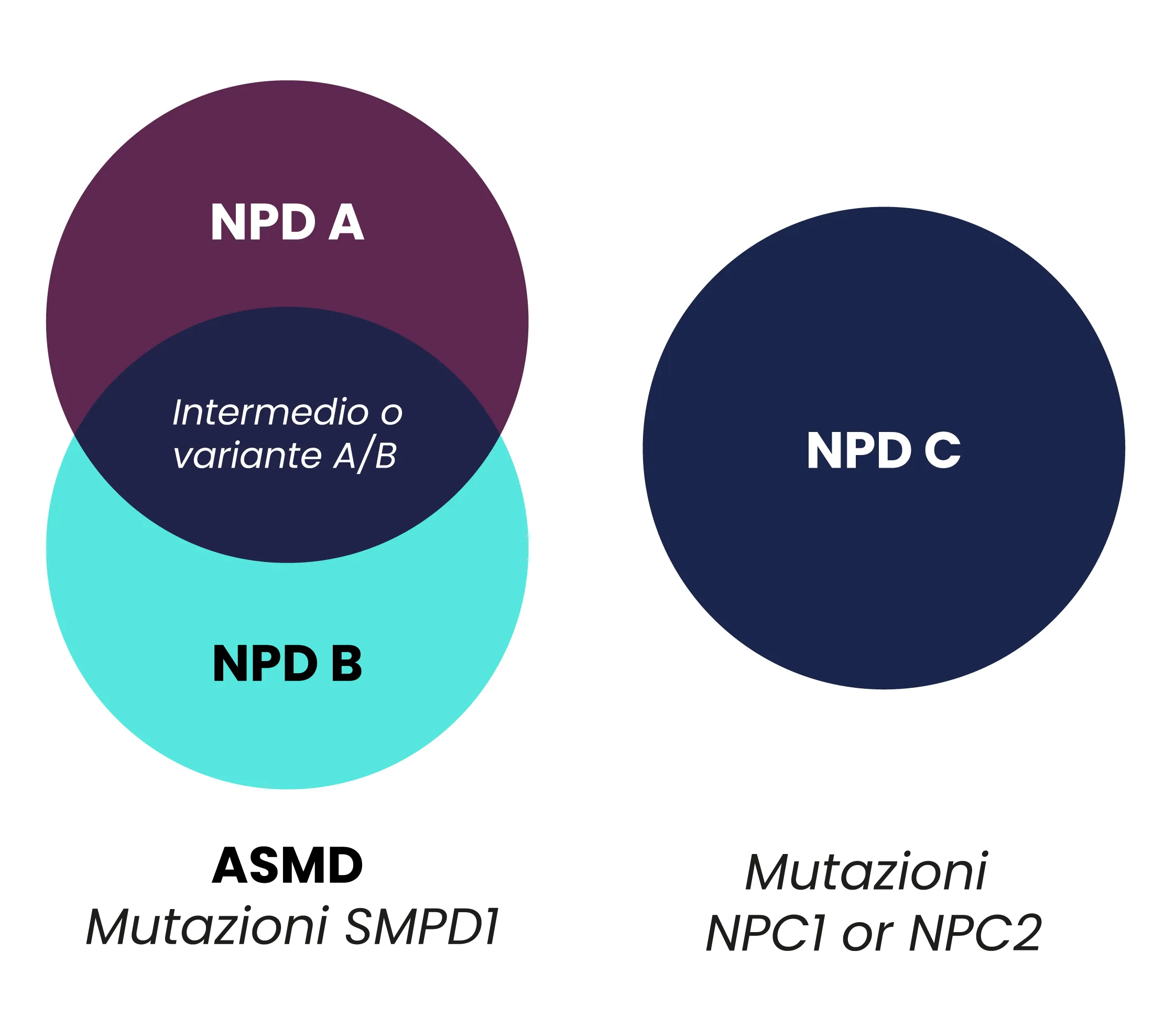
La NPD è storicamente classificata in 3 sottotipi: NPD A, NPD B, e NPD C.1
• I sottotipi A e B sono conosciuti anche come malattie da deficit della sfingomielinasi acida (o ASMD) e sono causati da mutazioni del gene sfingomielina fosfodiesterasi (SMPD1).2,3
• Il sottotipo C è associato a mutazioni dei geni NPC1 e NPC2, che causano difetti nel funzionamento di una delle due rispettive proteine deputate al trasporto del colesterolo all’interno di strutture cellulare dette lisosomi.2,4
_0.2022-06-24-16-12-34.webp)
ASMD
Il deficit di sfingomielinasi acida (ASMD) è una malattia genetica rara causata dall'alterazione di un enzima chiamato sfingomielinasi acida (ASM).
L'ASMD può insorgere sia nei bambini che negli adulti con sintomi iniziali lievi o severi. È una malattia cronica e potrebbe peggiorare nel tempo influendo sulla qualità di vita di chi la presenta.
Tipologie di ASMD
L'ASMD presenta 3 diversi tipi.
Approfondisci tutte le informazioni a riguardo perché potrebbero aiutarti a comunicare con il tuo medico e comprendere meglio i sintomi.
![]()
Tipo A:
La Malattia di Niemann-Pick tipo A insorge nella prima infanzia. Colpisce il cervello e il sistema nervoso, così come molti altri organi, e peggiora molto rapidamente.
![]()
Tipo A/B:
La Malattia di Niemann-Pick tipo A/B può insorgere durante l'infanzia. Colpisce più organi, incluso il cervello e il sistema nervoso, ma peggiora più lentamente rispetto al tipo A.
![]()
Tipo B:
La Malattia di Niemann-Pick tipo B può insorgere a qualsiasi età. Colpisce più organi, ma non il cervello, e peggiora più lentamente rispetto al tipo A. In alcuni casi, può presentare un coinvolgimento minimo del sistema nervoso centrale.
Scopri di più su come riconoscere i segni e i sintomi di ASMD
Leggi anche:
- Trasmissione ed ereditarietà di ASMD
Bibliografia
- Vanier MT. Chapter 176 - Niemann–Pick diseases. In: Olivier Dulac ML, Harvey BS, editors. Handbook of Clinical Neurology. Volume 113: Elsevier; 2013. p. 1717-21.
- Schuchman EH, Desnick RJ. Niemann-Pick disease types A and B: Acid sphingomyelinase deficiencies. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). New York, NY: McGraw-Hill. Chapt 144.
- Desnick JP et al. Mol Med. 2010;16:316-321.
- McGovern MM et al. Pediatrics. 2008;122(3):e341-e349.
Con il patrocinio di

MAT-IT-2100992