- Articolo
- Fonte: Campus Sanofi
- 18 gen 2024
Gestione della malattia
La Malattia di Pompe è una patologia autosomica recessiva, pertanto anche se i sintomi più comunemente riportati nella LOPD sono la debolezza dei muscoli prossimali e del tronco, l’intolleranza all’esercizio, la dispnea, il disturbo della tosse e le difficoltà nella deglutizione, si tratta di una malattia multisistemica, che può interessare diversi organi e sistemi.1
La cura dei pazienti e la gestione di questo disordine multisistemico devono essere affidate ad un team di medici multidisciplinare.2
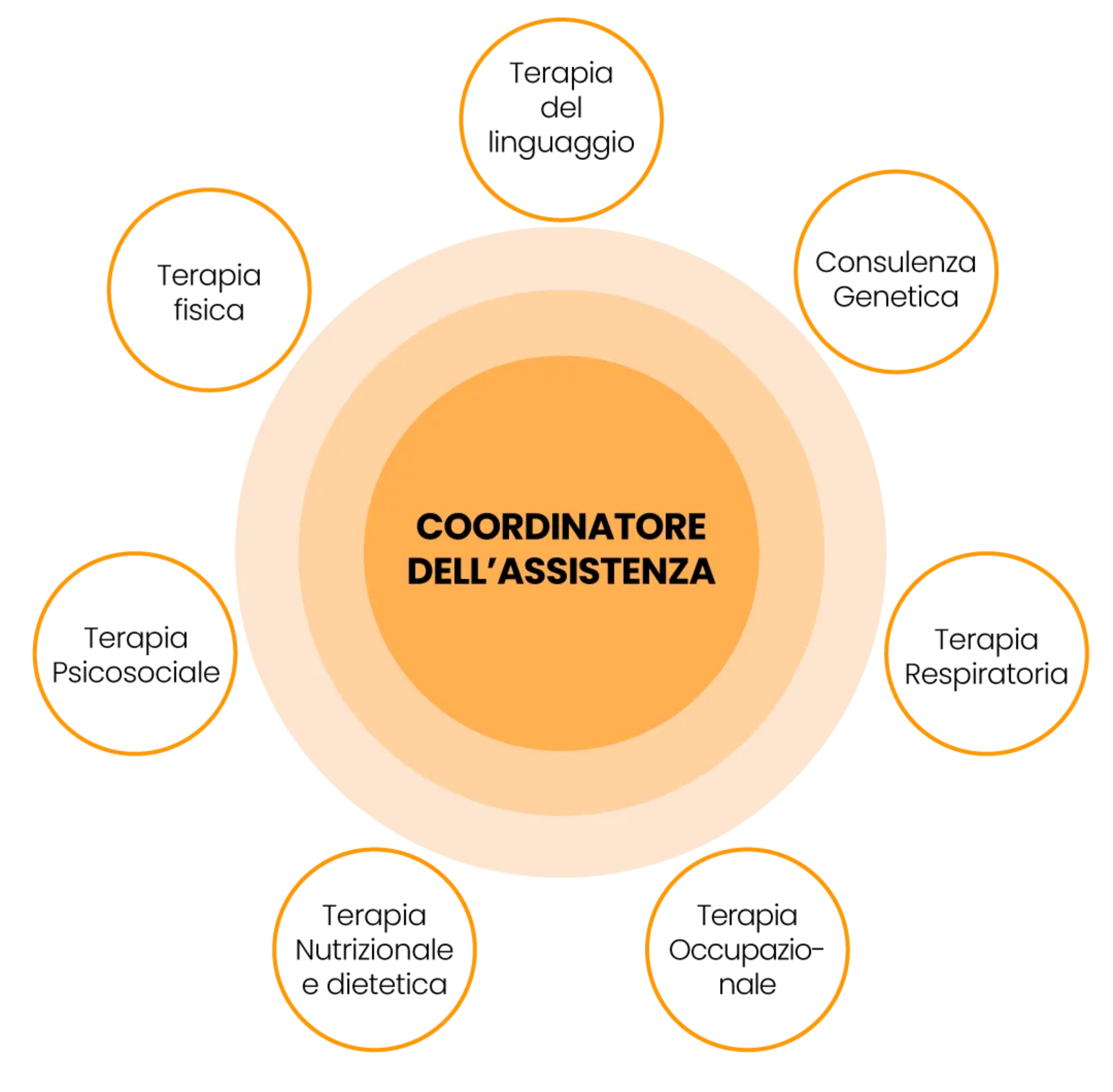
Nel settembre 2014 22 esperti nella gestione clinica dei pazienti con Malattia di Pompe di 9 paesi europei si sono riuniti a Naarden in un consorzio (EPOC: European Pompe Consortium) con l’obiettivo di:
- Stabilire un network europeo sulla Malattia di Pompe.
- Concordare un pannello minimo di variabili di outcome utili per la gestione clinica dei pazienti adulti con Malattia di Pompe.
- Dare indicazioni sui criteri di inizio e fine della terapia.
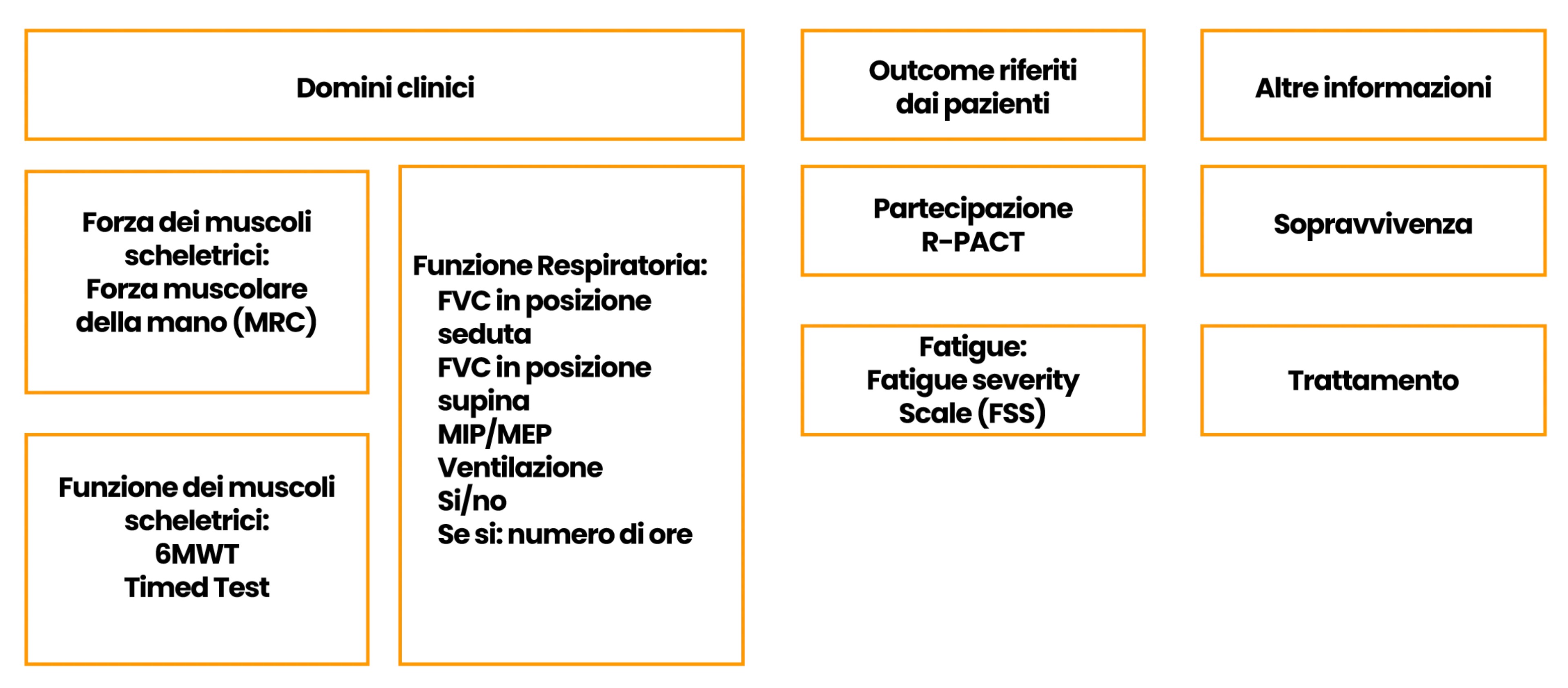
Il consorzio ha concordato le misure di outcome indispensabili per la condivisione all’interno del network e utili al follow up dei pazienti.
- Forza muscolare: MMT (Manual Muscle Testing) utilizzando la scala valutativa del MRC: estensori – flessori del collo; abduttori della spalla; flessori – estensori del gomito; flessori – estensori dell’anca; adduttori – abduttori dell’anca; flessori – estensori del ginocchio.
- Funzione muscolare: 6MWT (6 Minute Walking Test); GSGC (Gait: camminare per 10 metri; Stairs: salire 4 scalini; G: manovra di Gowers; C: alzarsi dalla sedia).
- Funzione respiratoria: test respiratori e stato ventilatorio.
- Outcome riferiti dai pazienti: test QoL e fatica.
- Altre informazioni: sopravvivenza e trattamento.
Monitoraggio costante
La natura progressiva della Malattia di Pompe, insieme alla natura imprevedibile di tale progressione, rendono fondamentale un monitoraggio regolare dei pazienti. Alterazioni delle condizioni cliniche dei pazienti possono essere identificate tempestivamente e il programma di trattamento/gestione può essere adattato conformemente.
Frequenza dei controlli:
- Tutti i pazienti con la forma infantile della Malattia di Pompe devono essere sottoposti ad un monitoraggio attento e frequente, vista la rapida progressione della malattia in questo gruppo di pazienti.4
- Nei pazienti con la forma ad esordio tardivo a progressione più lenta, si consiglia un controllo ogni 6-12 mesi.5,6
- Il team di medici che cura il paziente determinerà la natura e la frequenza delle valutazioni, in base alle necessità cliniche individuali.
Programma di valutazione del registro Pompe
Il registro Pompe ha sviluppato un programma di valutazioni, basato sulle raccomandazioni di specialisti internazionali con esperienza nel trattamento dei pazienti con Malattia di Pompe:
Le aree di valutazione chiave del monitoraggio delle manifestazioni e della progressione della Malattia di Pompe sono5-8
- Salute generale.
- Il monitoraggio del peso è importante in tutti i pazienti con Malattia di Pompe. Nei neonati, ad esempio, questa importanza deriva dalle difficoltà nell'alimentazione che possono rendere difficile il mantenimento di un peso corporeo normale. Sono disponibili anche diverse indagini e scale relative alla qualità della vita raccomandate per i pazienti, che possono supportare il monitoraggio della progressione della malattia e la valutazione del benessere mentale ed emotivo.
- Monitoraggio cardiaco: il monitoraggio cardiaco è indispensabile nei neonati con Malattia di Pompe. Infatti, la maggior parte dei neonati non trattati muore a causa di insufficienza cardiaca entro il primo anno di vita. RX del torace ed ECG consentono una valutazione costante della progressione della cardiomiopatia e aiutano a guidare le decisioni terapeutiche. Nei casi gravi, può essere utile un ECG ambulatoriale delle 24 ore per monitorare l'aumento del rischio di aritmia e di morte improvvisa.
- Monitoraggio delle vie respiratorie: l'insufficienza respiratoria è la causa più frequente di morte nei bambini e adulti con Malattia di Pompe anche fra quelli non sottoposti a ventilazione artificiale.7,8
L'insufficienza respiratoria non è sempre facile da riconoscere, pertanto test di routine della funzionalità polmonare (inclusa capacità vitale polmonare e resistenza diaframmatica) sono di importanza fondamentale, anche nei pazienti senza segni apparenti, in quanto un brusco peggioramento clinico può manifestarsi in qualsiasi momento. Anche un'anamnesi dettagliata del sonno, alla diagnosi e alle visite di controllo, può essere utile, in quanto i disturbi respiratori del sonno sono spesso una manifestazione precoce di patologie respiratorie.5
- Monitoraggio muscoloscheletrico e neurologico: valutazioni muscoloscheletriche e neurologiche, inclusi RX, elettromiografia (EMG) e traguardi motori comparativi aiutano a valutare e a tracciare le capacità funzionali e motorie dei pazienti, oltre alla progressione della malattia.5 Si tratta di strumenti importanti per decisioni informate sulla riabilitazione fisica e su altri interventi. Possono essere anche indicati test per verificare la densità minerale ossea, in quanto una diminuzione è frequente nei pazienti di diversi gruppi di età.
Gestione dei sintomi
L'impatto multisistemico della Malattia di Pompe è tale da dover suddividere la gestione dei sintomi e le cure di supporto in diverse categorie.
Il supporto respiratorio è una delle forme di gestione più critiche, essendo la maggior parte dei pazienti con Malattia di Pompe affetta da qualche forma di problema respiratorio, con l'insufficienza respiratoria come causa più frequente di morte prematura nei bambini e negli adulti affetti dalla malattia.6-8
Il supporto respiratorio spesso richiede:
- Uso di ossigeno supplementare (limitatamente a determinate situazioni).
- Eliminazione delle secrezioni delle vie aeree mediante tosse assistita e altre tecniche.
- Diverse forme di ventilazione meccanica per supportare i muscoli respiratori indeboliti, con necessità di tracheostomia nei casi più gravi.
- Cambiamenti dell'alimentazione per ridurre il rischio di aspirazione.
- Vaccinazioni aggiornate, incluse vaccinazioni anti-pneumococchi e influenza.
Altri interventi possono includere forme speciali di fisioterapia per rinforzare i muscoli respiratori indeboliti, 4 oltre ad una gestione aggressiva delle infezioni. In genere, con il progredire della malattia si rende necessario l'uso di un supporto respiratorio più frequente e invasivo.9,10
La Malattia di Pompe causa una degenerazione muscolare progressiva con diversi tipi e gradi di disabilità fisiche. La gestione di queste manifestazioni si focalizza sulla conservazione e sul miglioramento delle funzioni fisiche, oltre che sul miglioramento del comfort per il paziente. Uguale importanza rivestono il miglioramento o la prevenzione di complicanze secondarie, come contratture, deformità e bassa densità minerale ossea.5
I programmi di riabilitazione devono essere studiati su misura delle necessità individuali e possono includere diverse terapie e strategie, come:5,6
- Fisioterapia.
- Terapia occupazionale.
e di supporto.
Interventi e/o chirurgia ortopedica. - Logopedia.
- Dispositivi adattativi
In una recente pubblicazione11 sul ruolo della riabilitazione nei pazienti con LOPD, l'associazione italiana di Miologia (AIM), l'associazione degli Pneumologi Ospedalieri (AIPO), la società italiana di Neuroriabilitazione (SIRN) e la Società italiana di medicina fisica e riabilitazione (SIMFER), hanno formulato raccomandazioni su aspetti specifici per la Malattia di Pompe sulle procedure di riabilitazione in LOPD. In questo documento narrativo è stato esaminato il ruolo della riabilitazione nella gestione di LOPD e sono state definite le basi per i protocolli standardizzati per la valutazione e riabilitazione muscolo-scheletrica e i disturbi respiratori in pazienti con LOPD per soddisfare le esigenze nella gestione dei disturbi motori e della funzione respiratoria di questi pazienti.
I neonati spesso necessitano di una valutazione cardiaca e di una gestione dei sintomi più frequente. Questi pazienti sono a rischio di cardiomiopatia, cardiomegalia, insufficienza cardiaca congestizia, aritmie e arresto cardiaco durante gli interventi chirurgici.4
Il trattamento farmacologico deve basarsi sullo stadio della cardiomiopatia.5
La necessità di qualsiasi forma di intervento chirurgico nei neonati con Malattia di Pompe deve essere attentamente valutata rispetto ai rischi significativi dell'anestesia in questi pazienti. Pertanto, l'anestesia deve essere usata solo se assolutamente necessaria e deve sempre essere sorvegliata da un anestesista pediatrico e/o cardiaco esperto.
Trattandosi di una malattia cronica, degenerativa, con profondi risvolti sulla qualità della vita, la Malattia di Pompe può avere un considerevole impatto emotivo e psicologico sui pazienti e sulle loro famiglie. Pertanto, una parte integrante del trattamento dei pazienti consiste nel fornire diversi servizi di supporto psicosociale per alleviare il carico imposto dalla malattia e migliorare il benessere generale:5,6
- Consulenza individuale e famigliare.
- Accesso alla formazione sulla malattia.
- Organizzazioni di pazienti e gruppi di sostegno.
- Gruppi di supporto e reti per il collegamento con gli altri pazienti.
- Servizi di salute mentale e di assistenza sociale.
La debolezza muscolare associata alla Malattia di Pompe e i segni respiratori e cardiaci della malattia frequentemente possono compromettere lo stato di salute generale e il benessere dei pazienti.5,6
Ad esempio, molti pazienti hanno problemi di mobilità, sono più esposti alle infezioni e hanno difficoltà a mantenere un peso normale. Diverse terapie e strategie possono aiutare a gestire e controllare questi problemi:
- Terapia nutrizionale e altre tecniche per garantire un'alimentazione adeguata.
- Attenzione speciale ai farmaci e ai rispettivi effetti indesiderati, oltre che alle raccomandazioni post-intervento.
- Strategie per la prevenzione e la gestione delle infezioni.
Bibliografia
- Chan et al. Molecular Genetics and Metabolism 120 (2017) 163–172.
- Cupler EJ et al. Muscle Nerve. 2012 Mar; 45(3): 319–333.
- Schoser et al. Neuromuscular Disorders 25 (2015) 674–678.
- Kronn DF et al. Pediatrics 2017 Jul;140(Suppl 1):S24-S45.
- Kishnani PS et al. Genet Med 2006; 8:267-88.
- Cupler EJ et al. Muscle Nerve. 2012 Mar;45(3):319-33.
- Hirschhorn R, Reuser AJ. Glycogen Storage Disease Type II: Acid α-Glucosidase (Acid Maltase) Deficiency. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G., eds. The Online Metabolic and Molecular Bases of Inherited Disease. OMMBID.
- Winkel LP et al. J Neurol 2006; 252:875-84.
- Hagemans ML, Hop WC, Van Doorn PA, Reuser AJ, Van der Ploeg AT. Course of disability and respiratory function in untreated late-onset Pompe disease. Neurology 2006; 66:581-3.
- Mellies U, Lofaso F. Pompe disease: A neuromuscular disease with respiratory muscle involvement. Respir Med. 2009;103(4):477-84.
- Iolascon et al. Acta Myologica • 2018; XXXVII: p. 241-251.
MAT-IT-2100349