- Articolo
- Fonte: Campus Sanofi
- 26 dic 2023
Panoramica della Malattia di Gaucher

La malattia fu descritta per la prima volta dallo studente di medicina francese Philippe Gaucher nel 1882 mentre osservava una giovane donna affetta da splenomegalia e con delle caratteristiche cellule infarcite. Poco più di 50 anni dopo, il medico francese A. Aghion affermò che i pazienti che presentavano tale condizione accumulavano uno sfingolipide chiamato glucosilceramide. Ma fu solo nel 1965 che Roscoe Brady e i suoi collaboratori dimostrarono che la Malattia di Gaucher era causata dalla ridotta attività dell’enzima lisosomiale beta-glucosidasi acida.2
Il deficit dell'enzima glucocerebrosidasi è la causa della malattia: si verifica una compromissione del corretto metabolismo del glucocerebroside, che va quindi ad accumularsi nei lisosomi dei macrofagi, determinandone un aumento volumetrico e dando vita alle cosiddette "cellule di Gaucher". Queste si concentrano prevalentemente a livello della milza, del fegato e del midollo osseo, sebbene possano infiltrarsi anche in ulteriori tessuti, compresi il sistema linfatico e, più raramente, quello nervoso, alterandone le normali funzioni. L’accumulo di queste cellule in vari organi provoca una cascata di eventi fisiopatologici, tra cui la produzione di uno stato infiammatorio cronico e iper-metabolico.3
La Malattia di Gaucher è solitamente classificata in tre varianti fenotipiche in base all'assenza (tipo 1) o alla presenza (tipi 2 e 3) di manifestazioni neurologiche4:
- Il tipo 1 (non neuronopatico) presenta un'ampia variabilità in termini di manifestazioni cliniche, gravità e decorso, ma si distingue per l'assenza del coinvolgimento primario del sistema nervoso centrale.5 In alcune persone, i sintomi compaiono all'inizio dell'infanzia e peggiorano nel tempo, mentre in altri, i primi sintomi si osservano solo in età adulta.4
- Il tipo 2 (neuronopatico acuto) è solitamente evidente nei primi mesi di vita e include sintomi neurologici gravi. Questi bambini di solito non sopravvivono oltre i 2 anni di età.6
- Il tipo 3 (neuronopatico cronico) è caratterizzato da una malattia neurologica che progredisce lentamente e può essere confuso con la Malattia di tipo 1 nelle fasi iniziali. I pazienti che raggiungono l'adolescenza possono vivere fino all'età adulta.4
La classificazione dei diversi tipi di Malattia di Gaucher è utile per valutare le opzioni di gestione e come base per la consulenza genetica, ma si registra un crescente consenso nel considerare la malattia un continuum fenotipico caratterizzato da uno spettro di sintomi con manifestazioni neurologiche da lievi a gravi.7
Malattia di Gaucher - un continuum fenotipico
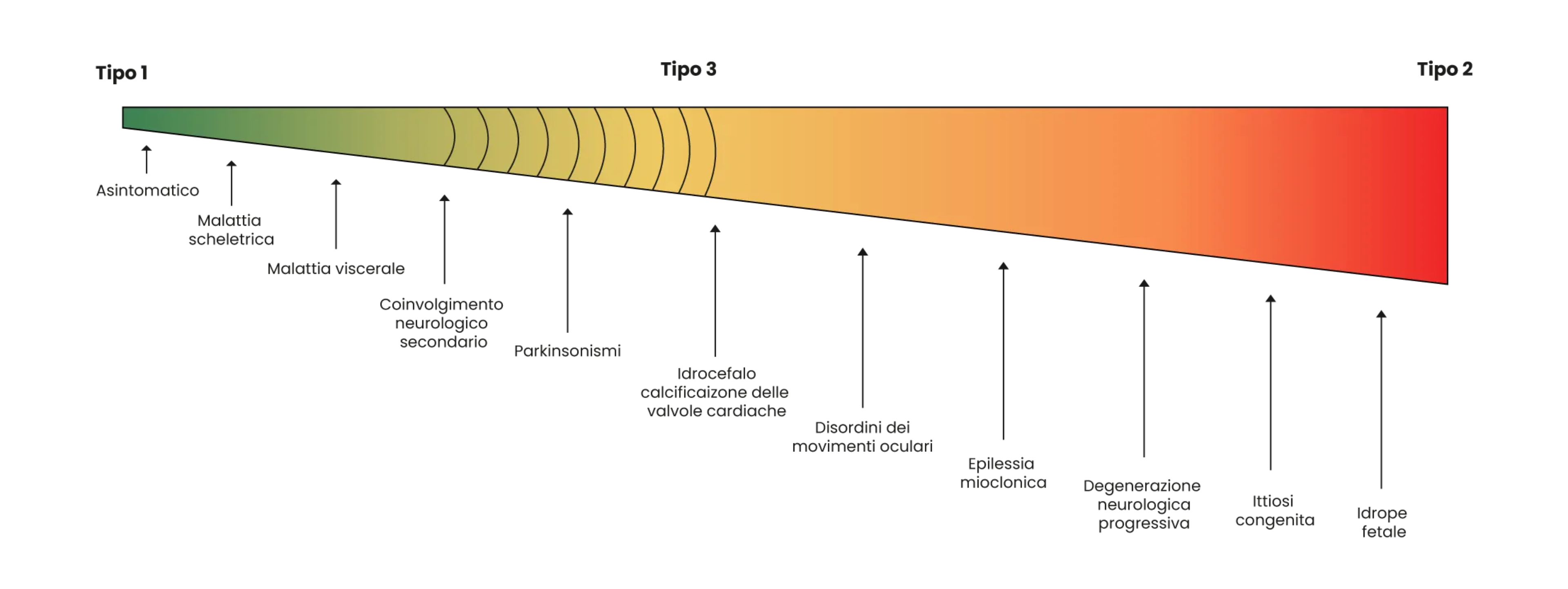
Immagine adattata da ref. 7.
Le categorie classiche dei tipi 1, 2 e 3 non sono nettamente distinguibili durante il decorso della malattia.
Leggi anche:
- Patogenesi
Bibliografia
- Mistry PK et al. Clin Adv Hematol Oncol. 2012;10:1-16.
- Brady RO. Baillieres Clin Haematol 1997;10(4):621-34.
- Grabowski G. Hematology Am Soc Hematol Educ Program 2012;1:13-18
- Grabowski GA, Petsko GA, Kolodny EH. Chapter 146: Gaucher disease. Valle D, Beaudet AL, Vogelstein B, et al, eds. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw Hill; 2013.
- Kaplan P et al. Eur J Pediatr (2013) 172:447–458.
- Gupta N et al. Blood Cells Mol Dis 2011;46(1):75-84.
- Sidransky E. Mol Genet Metab 2004;83(1-2):6-15.
MAT-IT-2100791